Biochemie
Anotace předmětu:
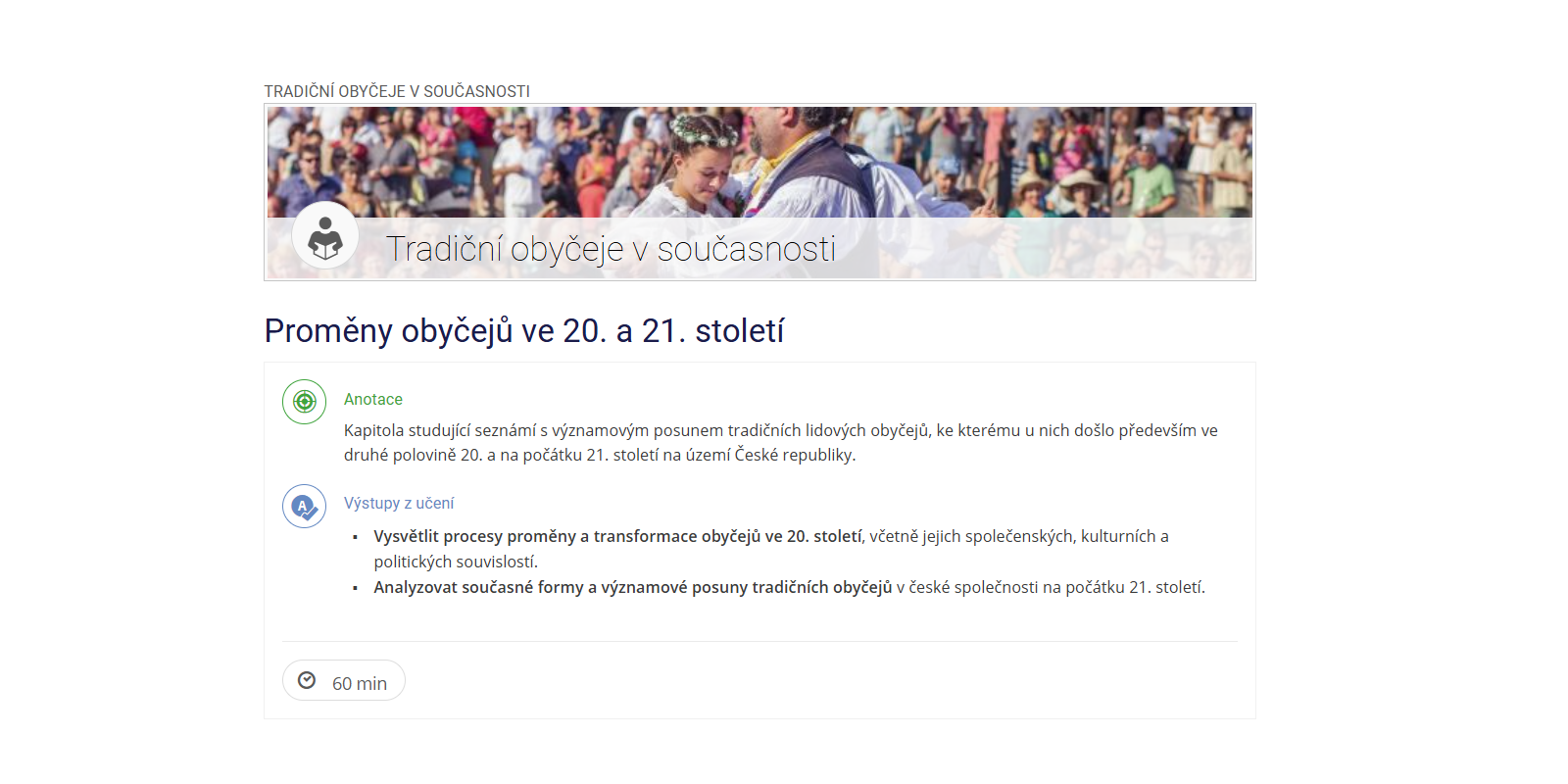
Předmět je koncipován jako teoreticko-praktický. Seznamuje se základními poznatky z oblasti biochemie. Informuje o biochemické rovnováze vnitřního prostředí organismu a jeho změnách v souvislosti s různými druhy onemocnění a poruch. Důležitou součástí jsou informace o metodách a technikách odběru biologického materiálu, jeho označování, uchovávání a odesílání ke zpracování do laboratoří. Seznamuje studenty s jednoduchými orientačními metodami biochemických vyšetření.
Kapitoly:
Kapitola obsahuje:
1
Odpovědník
13
Text
Kapitola obsahuje:
1
Obrázek
1
Odpovědník
8
Text
Kapitola obsahuje:
1
Odpovědník
12
Text
Kapitola obsahuje:
10
Obrázek
1
Odpovědník
3
Text
Kapitola obsahuje:
1
Odpovědník
6
Text
Kapitola obsahuje:
1
Odpovědník
4
Text
Kapitola obsahuje:
1
Odpovědník
4
Text
Kapitola obsahuje:
1
Odpovědník
3
Text
Kapitola obsahuje:
1
Odpovědník
6
Text
Kapitola obsahuje:
1
Odpovědník
* U některých testovacích otázek je více správných odpovědí, pro získání plného počtu bodů je potřeba zvolit všechny správné odpovědi.
Předchozí