Máte zapnutý náhled celé osnovy, zpět na běžné zobrazení.
Načítání a prohlížení osnovy může být v závislosti na množství obsahu pomalejší.
Hematologie
-
1 Laboratorní vyšetření krve
-
1.1 Červený krevní obraz
-
1.2 Bílý krevní obraz
-
1.3 Vyšetření hemostázy a hemokoagulace
-
1.3.1 Počet destiček
-
1.3.2 Doba krvácení (metoda podle Dukeho, Souliera a Ivyho)
-
1.3.3 Test rezistence kapilár (Rumpelův- Leedův test)
-
1.3.4 Test spotřeby protrombinu
-
1.3.5 Vyšetření PFA-100
-
1.3.6 Test agregace krevních destiček
-
1.3.7 Test retrakční schopnosti trombocytů
-
1.3.8 Systém plazmatických koagulačních faktorů
-
1.3.9 Přirozené inhibitory koagulačních faktorů
-
1.3.10 Vyšetření fibrinolytického systému
-
-
1.4 Laboratorní vyšetření v transfuzním lékařství
-
-
1 Anémie (onemocnění červených krvinek)
-
2 Poruchy koagulace
-
3 Nemaligní onemocnění
-
4 Hematologické malignance
-
5 Souhrnné testovací otázky
I Krev a její složky
1 Vlastnosti krve
1.1 Vlastnosti krve
|
Objem krve (l)
(% tělesné hmotnosti)
|
4,5–6
7–10
|
|
Objem krevní plazmy (l)
(% tělesné hmotnosti)
|
2,8–3,5
~ 5
|
|
Osmolalita plazmy (mOsm/l)
|
280–300
|
|
pH arteriální krve
|
7,4 ± 0,04
|
|
c (bílkoviny v plazmě)
(g/l)
|
64–82
|
|
c (albumin)
(g/l)
|
30–50
|
|
c (Na+)
(mmol/l)
|
136–148
|
|
c (Cl-)
(mmol/l)
|
95–110
|
|
c (HCO3-)
(mmol/l)
|
22–26
|
|
c (Ca2+)
(mmol/l)
|
2,15–2,61
|
|
c (glukózy)
mmol/l
|
3,9–5,6
|
|
c (cholesterol)
(mmol/l)
|
2,9–5,2
|
|
c (triacylglyceroly)
(mmol/l)
|
0,45–1,70
|
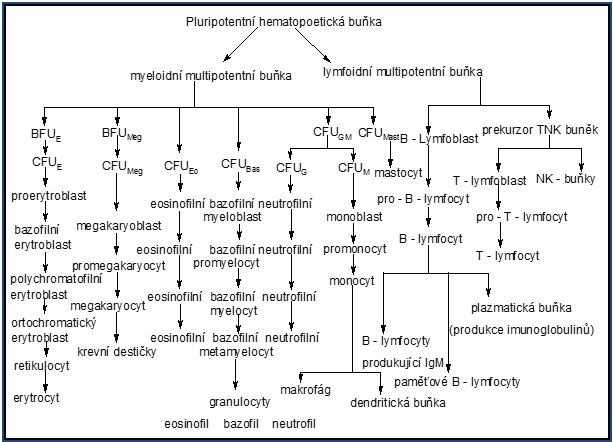
2 Krvetvorba (hematopoéza)
2.5 Testovací otázky

2.1 Vznik červených krvinek
|
Hematopoetické růstové faktory
|
Působení
|
|
Stem cell factor
(SCF, faktor kmenových buněk)
|
· sebeobnova pluripotentních kmenových buněk
· diferenciace na myeloidní a lymfatickou linii
|
|
Colony stimulating factor
(CSF, kolonie stimulující faktor)
|
· rozlišuje se několik typů podle linie, kde působí:
M – vývoj monocytů, stimulace fagocytózy; G – vývoj neutrofilů, stimulace fagocytózy; GM – vývoj všech CFU buněk z multipotentní buňky, stimulace fagocytózy;
|
|
Erytropoetin (Epo)
|
tvorba červených krvinek
|
|
Trombopoetin (TPO)
|
tvorba krevních destiček, nejdůležitější regulátor jejich tvorby
|
|
c-kit ligand (KL)
|
vývoj všech CFU buněk z multipotentní buňky, tvorba krevních destiček, vývoj a aktivace mastocytů
|
|
Flt-3 ligand (FL)
|
vývoj všech CFU buněk z multipotentní buňky
|
|
Leukemia inhibitory factor (LIF)
|
vývoj všech CFU buněk z multipotentní buňky
|
|
Oncostatin M (OSM)
|
vývoj všech CFU buněk z multipotentní buňky
|
|
Tumor necrosis faktor – α (TNF- α)
|
vývoj dendritických buněk (spolupráce s IL-3)
|
|
Interleukin-3 (IL-3)
|
zapojen do regulace všech linii krvetvorby s výjimkou vzniku T lymfocytů
|
|
Interleukin-5 (IL-5)
|
vývoj eosinofilů
|
|
Interleukin-6 (IL-6)
|
vývoj lymfocytů, zrání megakaryocytů
|
|
Interleukin-7 (IL-7)
|
vývoj B a T lymfocytů
|
2.2 Vznik krevních destiček
2.3 Vznik granulocytů
2.4 Vznik agranulocytů
3 Krevní složky
3.4 Testovací otázky
3.1 Červené krvinky
3.1.1 Červené krvinky
3.1.2 Hemoglobin
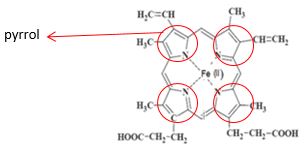
3.1.3 Metabolismus erytrocytů
3.1.4 Zánik červených krvinek
3.1.5 Sedimentace červených krvinek
3.2 Krevní destičky
3.2.1 Krevní destičky
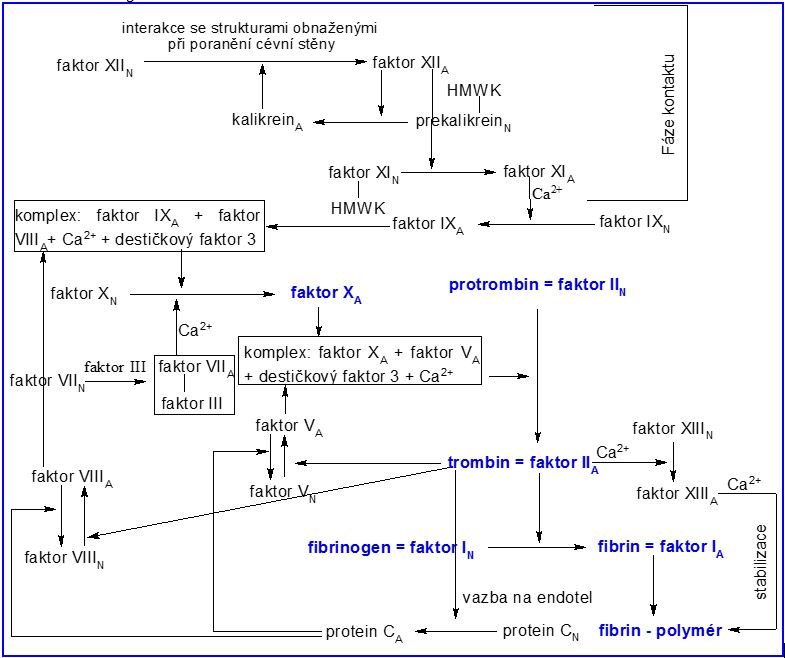
3.2.2 Hemostáza
|
Faktor
|
Charakteristika a působení
|
|
|
Faktor I
|
Fibrinogen
|
protein tvořený 3 polypeptidovými řetězci, v aktivní formě fibrin, který polymeruje a vytváří definitivní zátku
|
|
Faktor II
|
Protrombin
|
α2-globulin, v aktivní formě trombin – proteolytický enzym schopný aktivace fibrinogenu na fibrin, má mnoho dalších funkcí
|
|
Faktor III
|
Tkáňový faktor – tkáňový tromboplastin
|
transmembránový apoprotein, aktivuje faktor VII
|
|
Faktor IV
|
Ca2+
|
tvoří asi 50 % plazmatického Ca, účastní se většiny dějů v kaskádě
|
|
Faktor V
|
Proakcelerin
|
kofaktorem komplexu aktivátoru protrombinu
|
|
Faktor VII
|
Prokonvertin
|
součást komplexu aktivujícího faktor X
|
|
Faktor VIII
|
Antihemofilický faktor
|
součást komplexu aktivujícího faktor X; v plazmě cirkuluje navázaný na von Willebrandův faktor
|
|
Faktor IX
|
Christmasův faktor
|
proenzym, který se po své aktivaci účastní přeměny faktoru X
|
|
Faktor X
|
Stuartův-Prowerův faktor
|
důležitý v tvorbě protrombinového aktivátoru, v aktivní podobě enzym štěpící protrombin
|
|
Faktor XI
|
Plasma Thromboplastin Antecedent
|
proenzym kontaktního systému
|
|
Faktor XII
|
Hagemanův faktor
|
působí ve fázi kontaktu
|
|
Faktor XIII
|
Fibrin stabilizující faktor
|
podporuje tvorbu fibrinové sítě
|
|
Faktor XIV
|
Protein C
|
proenzym antikoagulační serin proteázy
|
|
Prekalikrein
|
součástí kontaktního systému
|
|
|
Kininogen o vysoké molekulové hmotnosti (HMWK)
|
kofaktorem ve fázi kontaktu
|
|
3.3 Bílé krvinky
3.3.1 Granulocyty
3.3.2 Agranulocyty
3.3.3 Imunita
4 Souhrnné testovací otázky
II Transfuzní lékařství
1 Krevní skupiny
1.4 Testovací otázky
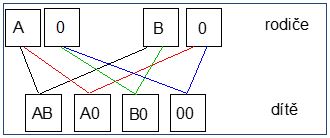
1.1 Systém AB0
|
Krevní skupina
|
Antigeny
|
Protilátky
|
Kompatibilní erytrocyty
|
|
|
H
|
anti-A, anti-B
|
0
|
|
A
|
A
|
anti-B
|
A, 0
|
|
B
|
B
|
anti-A
|
B, 0
|
|
AB
|
A, B
|
žádné
|
AB, A, B, 0
|
|
Rodič 1
|
Rodič 2
|
Dítě
|
|
A (A0)
|
B (B0)
|
A (A0), B (B0), 0 (00), AB (AB)
|
|
A (AA)
|
B (BB)
|
AB (AB)
|
|
A (AA)
|
B (B0)
|
A (A0), AB (AB)
|
|
A (A0)
|
B (BB)
|
B (B0), AB (AB)
|
|
A (A0)
|
0 (00)
|
A (A0), 0 (00)
|
|
B (B0)
|
0 (00)
|
B (B0), 0 (00)
|
|
A (A0)
|
A (A0)
|
A (AA), A (A0), 0 (00)
|
|
B (B0)
|
B (B0)
|
B (BB), B (B0), 0 (00)
|
|
AB (AB)
|
AB (AB)
|
A (AA), B (BB), AB (AB)
|
|
AB (AB)
|
0 (00)
|
A (A0), B (B0)
|
|
AB (AB)
|
A (A0)
|
A (AA), A (A0), B (B0), AB (AB)
|
|
AB (AB)
|
A (AA)
|
A (AA), AB (AB)
|
|
0 (00)
|
0 (00)
|
0 (00)
|
1.2 Rh systém
1.3 Ostatní skupinové systémy
1.3.1 Skupinový systém Kell
1.3.2 Skupinový systém MNS
1.3.3 Skupinový systém Duffy
1.3.4 Skupinový systém Lewis
1.3.5 Skupinový systém Lutheran
1.3.6 Skupinový systém Kidd
1.3.7 Skupinový systém Diego
2 Transfuze
2.6 Testovací otázky
2.1 Dárce a odběr krve a jejích složek
2.1.1 Kritéria výběru dárce
|
Věk
|
18-65 let
|
|
Hmotnost
|
≥ 50 kg
|
|
Hladina hemoglobinu
|
≥ 125 g/l (F), ≥ 135 g/l (M)
|
|
Hodnota hematokritu
|
≥ 0,380 (F), ≥ 0,400 (M)
|
|
Krevní tlak systolický/diastolický
|
≤ 180 mm Hg /≤ 100 mm Hg
|
|
Tepová frekvence
|
50-100 tepů/min
|
|
Celková bílkovina*
|
≥ 60 g/l
|
|
Trombocyty**
|
počet ≥150x109/l
|
|
Krevní skupina
|
Systém AB0 a RhD
|
2.1.2 Odběr krve a jejích složek
2.1.3 Zpracování odebrané krve a jejích složek
2.2 Transfuzní přípravky
2.2.1 Erytrocytární transfuzní přípravky
2.2.2 Trombocytové transfuzní přípravky
2.2.3 Transfuzní přípravky z plazmy
2.2.4 Granulocytární koncentráty
2.2.5 Bezpečnost TP
2.3 Krevní deriváty
2.4 Indikace podání transfuzních přípravků
2.4.1 Indikace transfuzních přípravků erytrocytů
|
Typ transfuzního přípravku
|
Indikace
|
|
Deleukotizované erytrocyty
|
a) riziko nehemolytické potransfuzní reakce
b) riziko imunizace buňkami jiného člověka (aloimunizace) před a po transplantacích (např. kostní dřeně, srdce, ledvin, jater)
c) riziko cytomegalovirové infekce (CMV) u CMV seronegativních příjemců a to před a po transplantaci orgánů, u pacientů s potlačenou imunitou, u nedonošených dětí a novorozenců, u těhotných žen, u dětí po operacích srdce a velkých cév
|
|
Ozářené erytrocyty
(pocházející od příbuzných)
|
snížená imunita
a) vrozené snížení imunity – imunodeficience
b) získané snížení imunity – po chemoterapii, či ozáření nebo po opakovaných transfuzích; před a po transplantaci kostní dřeně a orgánů
|
|
Promyté erytrocyty
|
výjimečně - u pacientů se známými protilátkami proti IgA nebo plazmatickým bílkovinám při těžkých alergických reakcích
|
2.4.2 Indikace transfuzních přípravků trombocytů
|
Příčina trombocytopenie
|
Indikace transfuze
|
|
Primární/sekundární selhání kostní dřeně
|
a) Terapeutická transfuze - indikována při aktivním krvácení
b) Profylaktická (prevence) transfuze - při poklesu počtu destiček pod hodnotu 5-10x109/l, i bez přítomnosti krvácení
|
|
Operace
a
invazivní výkony
|
a) Terapeutická transfuze – při hodnotách trombocytů:
< 50x109/l, u život ohrožujícího krvácení
< 100x109/l, u multiorgánového poranění či poranění CNS
b) Profylaktická transfuze – při hodnotách trombocytů:
< 10x109/l, před biopsií jater
< 20x109/l, před drobnými výkony (extrakce zubu, punkce kloubů, bronchoskopie, endoskopie GIT s biopsií tkáně, katetrizací)
< 50x109/l, před většími invazivními výkony (lumbální punkce, biopsie jater)
< 80x109/l, před epidurální anestezii
< 70-100x109/l, v průběhu neurochirurgických a očních operací
|
|
Autoimunitní
|
jen u život ohrožujících stavů
u plodu a novorozenců při hodnotě trombocytů < 30x109/l nebo 50x109/l společně s výskytem rizikových faktorů
|
2.4.3 Indikace plazmy
|
TP
|
Indikace transfuze
|
|
Plazma
|
a) při těžkém, život ohrožujícím krvácení nebo při přípravě na chirurgický výkon u pacientů s deficitem koagulačních faktorů, když není k dispozici chybějící koagulační faktor
b) při akutní diseminované intravaskulární koagulaci (vznik mnohačetných sraženin v cévách) spojené s krvácením
c) u trombotické trombocytopenické purpury (rozpad trombocytů)
d) při diagnostikovaném nedostatku koagulačních faktorů V, XI, XIII
e) při léčbě deficitu vitaminu K u novorozenců
f) u hemolytického onemocnění novorozenců v rámci výměnné transfuze
g) při masivních transfuzích nebo při hrazení masivních krevních ztrát koloidními a krystaloidními roztoky jako náhrada koagulačních faktorů
|
|
Ozářené plazmatické přípravky
|
ke snížení rizika potransfuzní reakce štěpu proti hostiteli
a) před a po transplantaci u pacientů se sníženou imunitou
b) u novorozenců a nedonošených dětí
c) u příjemců transfuzního přípravku od geneticky příbuzného dárce
|
2.4.4 Indikace transfuzních přípravků granulocytů
2.5 Potransfuzní reakce
|
Komplikace
|
Druh
|
Možné projevy
|
|
Neimunologické komplikace
|
Bakteriální infekce
(toxiny G- bakterií, kontaminace odběrových vaků, kontaminace při zpracování TP - hlavně u trombocytů)
|
Sepse (horečka > 39 °C, tachykardie >120 tepů/min. nebo zvýšení o 40 tepů/min., zvýšení nebo snížení systolického tlaku do 4 h po transfuzi)
|
|
Virová infekce
|
HIV,
hepatitida A, B, C;
cytomegalovirus;
vir Epstein-Barrové;
vir západonilské horečky; parvovir B19 (způsobuje útlum krvetvorby)
|
|
|
Parazitární infekce
|
malárie,
Chagasova choroba (v Evropě málo, spíše USA)
|
|
|
Prionová infekce
|
Creutzfeld-Jakobova choroba
(málo pravděpodobné)
|
|
|
Imunologické komplikace
|
Akutní hemolytické reakce (APHR)
(příčinou je inkompatibilita TP podle AB0 systému, výjimečně vysoký titr protilátek IgM)
|
tachykardie, teplota, třesavka, nevolnost, bolest na hrudi, zvracení, šok, pokles denní diurézy nebo úplné zastavení
|
|
Febrilní reakce
(vyvolána protilátkami proti
leukocytům a trombocytům v TP)
|
do 30-60 minut od podání transfuze
horečka, zimnice, zčervenání obličeje a hypotenze
|
|
|
Alergická reakce
(příčinou jsou protilátky IgE proti alergenům v TP)
|
teplota, bolest hlavy, kopřivka, dušnost, anafylaktický šok
|
3 Souhrnné testovací otázky
III Laboratorní diagnostika v hematologii
1 Laboratorní vyšetření krve
1.5 Testovací otázky
1.1 Červený krevní obraz
1.2 Bílý krevní obraz
|
KREVNÍ OBRAZ (KO)
|
|
|
Kvantitativní stanovení
Analyt (zkratka, jednotka)
|
Kvalitativní stanovení
Analyt (zkratka, jednotka)
|
|
Hemoglobin (Hb, g/l)
|
-
|
|
Hematokrit (HCT, HTC, l/l; %)
výpočet: (RBC.MCV)/10)
|
-
|
|
Počet erytrocytů (RBC, 1012/l)
|
Střední objem RBC (MCV, fl)
Průměrné množství Hb v RBC (MCH, pg)
Průměrná koncentrace Hb v RBC (MCHC, g/l)
Distribuční šíře velikosti RBC (RDW, %CV)
Retikulocyty (RET, 109/l, % při RET/RBC)
|
|
Počet krevních destiček (PLT, 109/l)
|
Střední objem trombocytů (MPV, fl)
Distribuční šíře velikosti trombocytů (PDW, specifické jednotky dle analyzátorů)
|
|
Počet bílých krvinek (WBC, 109/l)
|
-
|
|
Diferenciální rozpočet (WBC)
segmenty, tyče, eozinofily, bazofily, monocyty, lymfocyty, neutrofily
|
-
|
|
Parametr
|
Snížené hodnoty (změny počtu buněk)
|
Zvýšené hodnoty (změny počtu buněk)
|
Fyziologické hodnoty
|
|
Červené krvinky (RBC)
Hemoglobin (Hb)
Hematokrit (Ht, HCT, HTC)
|
Anémie
anémie, hemolytické reakce, hemoglinopatie, renální insuficience, krevní ztráty, aplázie dřeně, leukemie, lymfomy, těhotenství (při zvýšené hemodiluci)
|
polycytémie/polyglobulie
pobyt ve vyšších nadmořských výškách, po namáhavém tréninku, vrozená srdeční onemocnění, polycytémie, talasemie, stres, kouření, při těžkých formách dehydratace
|
RBC: M: 4,2-5,8 .1012/l
F: 3,8-5,2 .1012/l
Hb: M: 140-180 g/l
F: 120-160 g/l
HCT: M: 0,38-0,49
F: 0,35-0,46
(případně*100 v %)
|
|
Střední objem RBC (MCV)
|
anémie z nedostatku Fe, Zn, hemoglobinopatie (talasemie), anémie chronických onemocnění, sideroblastické anémie
|
nedostatek folátů, vit. B12 perniciózní anémie, alkoholismus, cirhóza jater, kouření
|
výpočet: HCT/RBC
obě pohlaví: 80-96 fl
|
|
Střední hmotnost Hb v erytrocytech (MCH)
Střední koncentrace Hb v erytrocytech (MCHC)
|
anémie z nedostatku Fe, Zn, hemoglobinopatie (talasemie), anémie chronických onemocnění, sideroblastické anémie
|
makrocytární anémie, dědičná sférocytóza
|
MCH:
výpočet: Hb/RBC
obě pohlaví: 27-32 pg
MCHC:
výpočet: Hb (g/dl)/HCT (%)
obě pohlaví: 0,32-0,37 g Hb/l ery
|
|
Počet retikulocytů
|
retikulopenie
neúčinná erytropoéza (aplastické anémie, anémie s poruchou vyzrávání červené řady), malignity
|
retikulocytóza
při zvýšené erytropoéze (akutní krevní ztráty, hemolytická anémie, talasemie)
|
0,025-0,075. 1012/l
|
|
Parametr
|
Snížené hodnoty (změny počtu buněk)
|
Zvýšené hodnoty (změny počtu buněk)
|
Fyziologické hodnoty
|
|
Počet leukocytů (WBC)
|
leukopenie
nedostatečná tvorba v kostní dřeni, malignity, nutriční nedostatek (Fe, B12,folát)
|
leukocytóza
infekce, stres, krvácení, malignity, fyzická námaha
|
4,4-10,5.109/l
|
|
Neutrofily
|
neutropenie
nutriční nedostatek, akutní leukemie, aplastická anémie, malárie, virové infekce (spalničky, zarděnky, hepatitida), lymfocytární leukemie, imunosuprese, léky (antibiotika, chemoterapeutika, analgetika)
|
neutrofilie
chronická a akutní myeloidní leukemie, myeloproliferace, zánětlivá onemocnění, infekce (bakteriální), léčba kortikosteroidy
|
2,5-7,5.109/l
|
|
Eozinofily
|
eozinopenie
šok, po léčbě kortikosteroidy
|
eozinofilie
parazitární infekce (škrkavky, hlísti), alergie, leukemie, ekzémy
|
0,04-0,11.109/l
|
|
Bazofily
|
-
|
chronická myeloidní leukemie, alergie
|
0,015-0,1.109/l
|
|
Lymfocyty
|
lymfocytopenie
AIDS, myelocytární leukemie, aplastická anémie, bakteriální infekce, lupus erythematodes
|
lymfocytóza
virová onemocnění, lymfocytární leukemie, Hodgkinova choroba
|
1,5-4,0.109/l
|
|
Monocyty
|
monocytopenie
porucha kostní dřeně, vlasatobuněčná leukémie, aplastická anémie, po léčbě Prednisonem
|
monocytóza
akutní infekce (virové, protozoární a parazitární), monocytární leukemie, maligní lymfomy, tuberkulóza, revmatoidní artritida
|
0,2-0,8.109/l
|
|
Trombocyty (PLT)
Střední objem trombocytů (PCV)
|
trombocytopenie
hypersplenizmus (zvětšená slezina), poškození dřeně, hemolytická anémie, imunitní poruchy (nejčastěji polékové), infekce (např. Helicobacterpylori), DIC, idiopatická imunitní trombocytopenická purpura, trombotická trombocytopenická purpura, heparinem indukovaná trombocytopenie
|
trombocytóza
nádorová onemocnění (GIT), zánětlivá střevní onemocnění, stavy po splenektomii, myeloproliferativní onemocnění (trombocytemie, polycythemia vera), infekce, po ztrátách krve, sideropenie, revmatoidní artritida, pankreatitidy.
|
150-450.109/l
7,8-11,0 fl
|
1.3 Vyšetření hemostázy a hemokoagulace
1.3.1 Počet destiček
1.3.2 Doba krvácení (metoda podle Dukeho, Souliera a Ivyho)
1.3.3 Test rezistence kapilár (Rumpelův- Leedův test)
1.3.4 Test spotřeby protrombinu
1.3.5 Vyšetření PFA-100
1.3.6 Test agregace krevních destiček
1.3.7 Test retrakční schopnosti trombocytů
1.3.8 Systém plazmatických koagulačních faktorů
1.3.9 Přirozené inhibitory koagulačních faktorů
1.3.10 Vyšetření fibrinolytického systému
1.4 Laboratorní vyšetření v transfuzním lékařství
1.4.1 Laboratorní vyšetření dárců krve a krevních složek
1.4.2 Předtransfuzní vyšetření
1.4.3 Kontrola transfuzních přípravků
IV Onemocnění krve
1 Anémie (onemocnění červených krvinek)
1.10 Testovací otázky
1.1 Klasifikace anémií
1.2 Sideropenická anémie
1.3 Sideroblastické anémie
|
|
dědičné
|
získané
|
|
etiopatogeneze
|
defekty enzymů účastnících se tvorby hemoglobinu
|
a) reverzibilní – jsou-li odstraněny příčiny
b) přetrvávající – součást jiného onemocnění
|
|
známo u d-aminolevulinátsyntetázy s koenzymem pyridoxalfosfát
|
||
|
nedostatek pyridoxinu
|
||
|
alkoholismus
|
||
|
léky
|
||
|
otrava olovem
|
||
|
dědičnost
|
různé formy – autozomální dominantní i recesivní, někdy na chromozomu X
|
-
|
|
příznaky
|
hepatomegalie
|
-
|
|
splenomegalie
|
||
|
srdeční anémie
|
||
|
obecné příznaky anémie
|
||
|
krevní obraz
|
mikrocytární anémie
|
normocytární až makrocytární anémie
|
|
hypochromní anémie
|
||
|
zpožděné zrání cytoplazmy u prekurzorů
|
||
|
¯ plazmatické železo
|
||
|
léčba
|
pyridoxin
|
odstranění příčiny nemoci
|
|
chelatační léčba (odstranění nadbytku železa)
|
pyridoxin
|
1.4 Aplastické anémie
|
typ anémie
|
vrozené
|
získané
|
|||
|
Fanconiho
|
Blackfanova-Diamondova
|
Vrozená diskeratóza
|
Aplazie erytropoézy
|
Aplastická anémie
|
|
|
dědičnost
|
autozomálně-recesivní
|
autozomálně
|
podle daného genu
|
-
|
-
|
|
patogeneze
|
zapojeno 13 genů
|
není přesně známa
|
zapojeny 4 geny
|
imunitní reakce
|
toxické vlivy/imunitní reakce
|
|
důsledek
|
apoptóza kmenových a progenitorových buněk
|
aplazie nebo hypoplazie erytropoézy
|
zkracování telomer Þ ¯ proliferativní potenciál
|
zástava erytropoézy
|
selhání proliferace/diferenciace kmenových buněk
|
|
klinické nálezy
|
hnědá pigmentace kůže
|
malý vzrůst
|
retikulární pigmentace kůže
|
|
acyklické krvácení u žen
|
|
kostní abnormality
|
krátký krk
|
dysplastické nehty
|
krvácení z nosu
|
||
|
dislokace kyčlí
|
hepatomegalie
|
mikrocefalie
|
petechie na kůži
|
||
|
endokrinní odchylky
|
splenomegalie
|
osteoporóza
|
slabost
|
||
|
mikrooftalmie
|
rozštěp patra
|
aseptické nekrózy
|
únava
|
||
|
mentální retardace
|
rozštěp rtu
|
leukoplakie
|
nevýkonnost
|
||
|
riziko vzniku nádorů
|
vysoké
|
vysoké
|
cca 1/3 nemocných
|
u žen
|
u imunosupresivní léčby
|
|
leukémie
gynekologické nádory
nádory mozku
myelodysplazie
|
leukémie
myelodysplazie osteosarkom
|
leukémie
myelodysplazie dlaždicobuněčné karcinomy
|
tymom
|
myelodysplazie
|
|
|
krevní obraz
|
¯ hemoglobin
|
¯ retikulocytů
|
pancytopenie
|
¯ retikulocytů
|
pancytopenie
makrocytární anémie
|
|
trombocytopenie
|
erytropoetinu
|
normocytární anémie
|
|||
|
leukopenie
|
adenozin-deamináza
|
normochromní anémie
|
|||
|
normocytární anémie
|
makrocytární anémie
|
v dřeni chybí erytrocyty
|
|||
|
fetální hemoglobin
|
fetální hemoglobin
|
erytropoetinu
|
|||
|
léčba
|
transplantace kostní dřeně
|
transplantace kostní dřeně
|
transplantace kostní dřeně
|
kortikosteroidy
|
záleží na závažnosti
anabolika
|
|
kortikosteroidy
|
androgeny
|
imunosupresivní
|
|||
1.5 Talasemie
|
|
α-talasemie
|
β-talasemie
|
|
patogeneze
|
delece nebo mutace genů (2 na 16. chromozomu) pro α-globin
|
bodové mutace ovlivňující
regulaci nebo expresi (případně delece) genu pro β-globin na 11. chromozomu
|
|
důsledek
|
žádná (homozygoti α0)/ nedostatečná syntéza α-globinu
|
žádná (homozygoti β0)/nedostatečná syntéza β-globinu
|
β-globin je nadále syntetizován =>
tvorba hemoglobinu H složeného ze
4 β-řetězců => nestabilní |
α-globin je nadále syntetizován =>
precipitace přebytečných α-řetězců => poškození erytrocytární membrány => hemolýza
|
|
|
méně stabilní hemoglobin =>
podléhá autooxidaci – uvolňuje železo
|
||
|
porušena i erytropoéza
|
||
|
typy
|
a) tiché nosičství (αα, α-)
b) α-talasemie minor (α-, α-) nebo (--, αα)
c) choroba hemoglobinu H (α-, --)
d) choroba hemglobinu Barts (--, --) tedy α0
|
a) β-talasemie minor (ββ+) nebo (ββ0)
b) β-talasemie intermedia
c) β-talasemie major (β0β0, β0β+, β+β+)
|
1.6 Megaloblastové anémie
1.7 Korpuskulární hemolytické anémie
1.7.1 Poruchy membrány erytrocytů
|
Typ anémie
|
Hereditární sférocytóza
|
Hereditární eliptocytóza
|
Hereditární stomatocytóza
|
Hereditární akantocytóza
|
Paroxyzmální noční hemoglobinurie
|
|
dědičnost
|
autozomálně dominantně
|
autozomálně dominantně
|
autozomálně dominantně
|
autozomálně recesivní
|
získaná anémie
|
|
incidence
|
1: 1000 až 1:2500
|
1: 2000 až 1:4000
|
|
cca 100 případů
|
2-5:1 000 000
|
|
příčiny
|
mutace genu SPTA1
|
molekulární poruchy spektrinu
|
nedostatek stomatinu
|
defektní syntéza apoB
|
mutace genu fosfatydylinozitolglykanu A na chromozomu X
|
|
mutace genu ANK1
|
deficit glykoforinu C
deficit proteinu 4.1
|
změny v genu pro xerocytózu
|
|||
|
důsledek
|
porucha formování cytoskeletu erytrocytu
|
oslabení či fragilita membránového skeletu erytrocytu
|
zvýšená permeabilita membrány
|
chybění frakcí lipoprotinů: VLDL, LDL, chylomikra
|
intravaskulární hemolýza
|
|
nedostatek spektrinu, případně ankyrinu a pallidinu
|
změny objemu erytrocytů -
hyperhydratace, ¯ xerocytóza
|
větší množství sfingomyelinu v membráně
|
defekt bílkovin buněčné membrány podílejících se na útlumu aktivace komplementu
|
||
|
nestabilita lipidové dvojvrstvy membrány
|
erytrocyty mají eliptický tvar
|
krvinky ve tvaru akantocytů (ostnité erytrocyty)
|
|||
|
přeměna ve sférocyt
|
změna hladiny sodíku a draslíku v buňce
|
||||
|
klinické příznaky
|
anémie
žloutenka
splenomegálie
hyperplázie erytropoézy
|
splenomegálie
|
dost často asymptomatický průběh
|
poruchy růstu
mentální retardace
steatorea
progresivní ataxie
|
sklony k trombóze
náchylnost k infekcím
hemoglobinurie po spánku
|
|
krevní obraz
|
normocytární anémie
|
bez anémie až po těžkou anémii
|
v nátěrech stomatocyty
|
normochromní anémie
|
membránový defekt trombocytů
|
|
normochromní anémie
|
retikulocytů
|
||||
|
retikulocytů
|
retikulocytů
|
v nátěrech akantocyty
|
¯ granulocytů
|
||
|
v nátěrech sférocyty
|
v nátěrech eliptocyty
|
normocytární anémie
|
|||
|
¯ osmotická rezistence
|
autohemolýza
|
normochromní anémie
|
|||
|
léčba
|
Splenektomie
|
splenektomie
(u těžké formy)
|
splenektomie
(je nutná jen zřídka)
|
úmrtí mezi 20. a 30. rokem života
|
transplantace kostní dřeně
protilátka proti C5 komplementu
|
1.7.2 Poruchy struktury hemoglobinu
1.7.3 Poruchy metabolismu erytrocytů
1.8 Extrakorpuskulární hemolytické anémie
1.8.1 Aloimunitní hemolytické anémie
1.8.2 Autoimunitní hemolytické anémie
|
Typ anémie
|
AHA s tepelnými protilátkami
|
Syndrom chladové hemolýzy
|
Polékové AHA
|
Symptomatické AHA
|
|
|
Choroba chladových aglutininů
|
Paroxyzmální chladová hemoglobinurie
|
||||
|
Charakteristika autoprotilátek
|
optimálně působí při 37 °C
|
reagují při teplotách pod 32 °C
|
léky indukovaná hemolýza – známo asi 100 léků, které mohou takto působit
|
tvorba autoprotilátek je spojena s jiným základním onemocněním – nádorová, revmatická, infekční, autoimunitní, imunitní onemocnění
|
|
|
namířeny nejčastěji proti antigenům Rh systému
|
protilátky třídy IgM (vzácně IgG) se schopností aktivovat komplement – intravaskulární hemolýza
|
||||
|
protilátky třídy IgG – vedoucí k extravaskulární hemolýze
|
Děti: polyklonální protilátky
Starší lidé:monoklonální protilátky
|
tzv. Donathova-Landsteinerova protilátka; třídy IgG; specifita: anti-P
|
|||
|
Klinické příznaky
|
slabost, únavnost, dušnost
|
bledost, žloutenka
|
rychle vznikající nemoc
|
existují různé typy:
a) lék přímo ovlivňuje imunitní systém – tvorba protilátek
b) lék se váže na membránu krvinek a antigen reaguje s tímto komplexem
c) autoprotilátky reagují, je-li ve směsi lék i membrána krvinek - imunokomplexový typ
|
podmíněno základním onemocněním
|
|
nevysvětlitelná horečka
|
cyanotické zbarvení terminálních částí těla (prsty, nos, ušní boltce)
|
horečka
|
|||
|
bolesti břicha
|
bolesti zad a končetin
|
||||
|
nechutenství
|
nauzea a zvracení
|
||||
|
zmatenost
|
akrocyanóza (symetrické modrofialové zbarvení rukou)
|
bolesti břicha a hlavy
|
|||
|
splenomegalie
|
rudá až černá moč
|
||||
|
hepatomegalie
|
mírná žloutenka
|
||||
|
Krevní obraz
|
retikulocytů
|
retikulocytů
|
hemoglobinemie
|
||
|
leukocytóza a neutrofilie
|
leukocyty v normě
|
hemoglobinurie
|
|||
|
normocytová/makrocytová anémie
|
v nátěrech patrná autoaglutinace erytrocytů
|
methemalbumin
|
|||
|
v nátěrech sférocyty, anizocytóza
|
změna barvitelnosti erytrocytů
|
v akutní fázi ¯ retikulocyty
|
|||
|
normální či zásoby železa
|
při obnově retikulocyty
|
||||
|
Léčba
|
primárně: glukokortikoidy
|
kortikosteroidy
|
kortikosteroidy
|
||
|
následně: splenektomie či imunosupresiva
|
rituximab v kombinaci s cyklofosfamidem
|
zabránit prochlazení nemocného
|
|||
1.8.3 Neimunitní hemolytické anémie
1.9 Akutní posthemoragická anémie
2 Poruchy koagulace
2.5 Testovací otázky
2.1 Poruchy primární hemostázy
2.1.1 Trombocytopenie
|
rozdělení dle typu onemocnění
|
onemocnění
|
|
vrozené choroby
|
Fanconiho anémie
|
|
familiární amegakaryocytární trombocytopenie
|
|
|
Wiskottův-Aldrichův syndrom
|
|
|
Mayova-Hegglinova anomálie
|
|
|
Bernardův-Soulierův syndrom
|
|
|
Paris-Trousseau (Jacobsenův) syndrom
|
|
|
imunologicky podmíněné choroby
|
idiopatická trombocytopenická purpura
|
|
potransfuzní purpura
|
|
|
izoimunitní novorozenecká trombocytopenie
|
|
|
Evansův syndrom
|
|
|
polékové trombocytopenie
|
|
|
heparinem indukovaná trombocytopenie
|
|
|
HIV-asociovaná trombocytopenie
|
|
|
trombocytopenie z nedostatečné tvorby destiček
|
Megaloblastové anémie
|
|
v důsledku užívání některých léků
|
|
|
v důsledku užívání alkoholu
|
|
|
v důsledku nedostatku železa
|
|
| paroxyzmální noční hemoglobinurie
|
|
|
trombocytopenie ze zvýšeného zániku destiček
|
Trombotické mikroangiopatie
|
|
Syndrom Kasabachův–Merrittové
|
|
|
Diseminovaná intravaskulární koagulace
|
|
|
trombocytopenie z abnormální redistribuce destiček
|
onemocnění sleziny
|
|
Hypotermie
|
|
|
diluční trombocytopenie
|
2.1.2 Trombocytóza
|
Typ onemocnění
|
Onemocnění
|
|
bakteriální infekce
|
pneumonie
|
|
pyelonefritida
|
|
|
osteomyelitida
|
|
|
hnisavé artritidy
|
|
|
chronické záněty jizev
|
|
|
plicní tuberkulóza
|
|
|
zánětlivá onemocnění
|
revmatoidní artritida
|
|
vaskulittida
|
|
|
nefritida
|
|
|
cirhóza
|
|
|
nádorová onemocnění
|
plicní nádory
|
|
až u 50% solidních nádorů
|
|
|
ostatní příčiny
|
stav po splenektomii
|
|
nedostatek železa
|
|
|
krvácení či hemolýza
|
|
|
léčba megaloblastové anémie
|
|
|
stres
|
2.1.3 Trombocytopatie
Typ poruchy | Onemocnění | |
vrozené | porucha adhezivity destiček | |
porucha agregability destiček | Glanzmannova-Naegeliho tromboastenie | |
porucha sekrece destiček – porucha skladovacích granulí a uvolňovací reakce | Heřmanského-Pudlákův syndrom | |
Syndrom šedých destiček | ||
„aspirin like disease“ | ||
porucha prokoagulační aktivity destiček | Scottův syndrom | |
získané | trombocytopatie provázející jiná onemocnění | |
paraproteinemie | ||
renální insuficience | ||
jaterní onemocnění | ||
autoimunitní choroby | ||
polékové trombocytopatie | ||
2.2 Vrozené krvácivé stavy
2.2.1 Hemofilie
2.2.2 Von Willebrandova choroba
2.2.3 Ostatní vrozené krvácivé stavy
2.3 Získané poruchy krevního srážení
|
Příčina
|
Působení
|
Projevy
|
|
|
Nedostatek vitaminu K
|
HON;
podávání antibiotik;
parenterální výživa;
porušená absorpce vitaminu K;
podávání antagonistů vitaminu K
|
vitamin K katalyzuje karboxylaci zbytků kyseliny γ-glutamové
- ovlivňuje syntézu faktorů koagulace II, VII, IX, X a proteinů C, S, Z
|
náchylnost ke krvácení
|
|
Jaterní
postižení
|
poruchy cévní stěny;
poruchy primární hemostázy;
nedostatek koagulačních faktorů;
zvýšená fibrinolytická aktivita;
dysproteinemie (porušená funkce proteinů nutných pro srážení);
vznik protilátek proti faktorům
|
játra jsou místem syntézy a i eliminace většiny faktorů koagulace – ovlivněny jsou jak srážlivé tak protisrážlivé funkce
|
krvácivé projevy
|
|
Uremie
|
cévní abnormality;
defekt krevních destiček;
porucha metabolismu buněk cévního endotelu;
zvýšená hladina antitrombinu;
zvýšená hladina faktoru VIII;
zvýšená hladina fibrinogenu;
snížená fibrinolytická aktivita;
snížení aktivity proteinu C
|
zvýšená hladina zánětlivých cytokinů a proteinů akutní fáze;
porucha koagulačního i fibrinolytického systému
|
abnormální krvácení – nejčastěji do GIT
|
|
Nádorová onemocnění
|
vlastní nádorové onemocnění
- prorůstání nádorů do cév
- útlak cév nádory
- aktivace krevních destiček
- vylučování nádorového prokoagulans
- exprese tkáňového faktoru
- indukce cytokinů
vliv léčby
postižení jater či dalších orgánů
|
kvantitativní i kvalitativní změny trombocytů;
aktivace fibrinolýzy;
rozvoj DIC;
aktivace a defekt plazmatických faktorů
|
solidní nádory – sklon k trombóze; leukémie – krvácivé projevy
|
|
Paraproteinemie
|
výskyt patologických proteinů – paraproteinů
|
paraproteiny mohou inhibovat působení a funkčnost destiček a koagulačních faktorů;
působit jako protilátky;
ukládat se v podobě amyloidu
|
krvácivé projevy
|
|
Trauma
|
poranění a masivní krvácení;
hypotenze;
snížená saturace tkání kyslíkem;
hemolýza;
podávání náhradních roztoků či masivních transfuzí;
|
hypotermie; acidóza; poruchy hemostázy; mikrovaskulární krvácení, rozvoj DIC
aktivace fibrinolýzy;
diluční koagulopatie
|
masivní krvácení
|
|
Sepse
|
infekce, sepse, septický šok –
systémová odpověď organismu na infekci
|
poškození endotelu; aktivace koagulace; inhibice fibrinolýzy; snížená aktivita inhibitorů krevního srážení;
rozvoj DIC
|
|
|
Hadí jedy
|
směs biologicky aktivních proteinů a peptidů (většinou toxických)
|
pro krevní destičky i pro všechny koagulační faktory je znám nějaký hadí jed, který ovlivňuje jejich funkci
|
|
2.4 Trombotické stavy
2.4.1 Trombofilie
2.4.2 Trombotické mikroangiopatie
3 Nemaligní onemocnění
3.3 Testovací otázky
3.1 Kvalitativní poruchy leukocytárního systému
3.1.1 Morfologické anomálie leukocytů
|
Anomálie
|
Příčina
|
Porucha
|
Příznaky/projevy
|
|
vrozené morfologické anomálie
|
|||
|
Pelgerova-
Huëtova
|
autozomálně dominantně dědičná mutace genu pro lamin B receptor
|
neschopnost segmentovat jádra granulocytů
|
heterozygoti – funkce neutrofilů neovlivněna; homozygoti - edémy u plodu, Greenbergův syndrom, dysplázie
|
|
Alderova-
Reillyho
|
autozomálně recesivně dědičná porucha
|
skladování mukopolysacharidů v buňce, v podobě granul
|
|
|
hypersegmentace
jader granulocytů
|
autozomálně dominantně dědičná porucha
|
nadměrná segmentace jader granulocytů
|
bez klinických důsledků
|
|
Mayova-
Hegglinova
|
autozomálně dominantně dědičná mutace genu MYH9
|
Döhleho inkluze v neutrofilech
|
leukopenie, trombocytopenie, obrovské trombocyty
|
|
Jordanova
|
autozomálně recesivně dědičná porucha
|
přítomnost vakuol v neutrofilech, monocytech
|
svalová dystrofie, ichtyóza (porucha rohovatění kůže)
|
|
získané morfologické anomálie
|
|||
|
Pseudo-Pelgerovy
změny
|
provázejí jiná onemocnění: infekce, leukémie, metastázy karcinomů kostní dřeně;
léky: kolchicin, sulfonamid
|
|
|
|
Döhleho
inkluze
|
provázejí jiná onemocnění: infekce, popáleniny, traumata, malignity; v těhotenství
léky: cytostatika
|
cystická tělíska u neutrofilních granul
|
|
|
Toxická granulace
a vakuolizace
|
provázejí jiná onemocnění: infekce, septické stavy
|
zhrubělá granulace a masivní vakuolizace
|
|
3.1.2 Funkční změny leukocytů
3.2 Kvantitativní poruchy leukocytárního systému
|
|
NADBYTEK
|
NEDOSTATEK
|
|
|
Neutrofily
|
název
|
neutrofilie
|
neutropenie
|
|
referenční hodnota
|
> 7,5*109 neutrofilů/l (pro dospělé)
|
< 1,5*109 neutrofilů/l (pro dospělé)
|
|
|
příčiny vzniku
|
- těhotenství, obezita, sportovní výkon
- emocionální vlivy (strach, panika, úzkost)
- infekce, záněty
- tumory (karcinomy, lymfomy)
- léky (lithium, heparin, digitalis)
- toxiny (endotoxin, hadí jedy)
- chemické látky a hormony (histamin, acetylcholin, serotonin, cystein, olovo, adrenalin, noradrenalin, kortikosteroidy)
- jiná onemocnění (krvácení, hemolytická anémie, leukémie, trombocytopenie, chronická idiopatická leukocytóza, granulomatózní nemoc, dna, eklampsie, uremie, diabetická acidóza)
- potransfuzní reakce
|
a) vrozená
- důsledek defektu kmenové buňky krvetvorby
- porucha dělení a zrání během granulopoézy (chronická benigní neutropenie)
- vrozená agranulocytóza – tzv. Kostmannův syndrom
b) získaná
- poruchy na úrovni mateřské buňky – získaná aplázie či hypoplázie kostní dřeně, leukémie
- virové infekce – viry hepatitidy A, B, chřipky, spalničky, zarděnky, neštovice, infekční mononukleóza
- bakteriální infekce – salmonelózy, TBC, tularemie
- po podání některých léčiv – peniciliny, sulfonamidy, tyreostatika, antirevmatika, antipyretika, cytostatika
c) autoimunitní
- revmatoidní artritida
|
|
|
příznaky
|
dány onemocněním, které neutrofilie provází
|
infekce (záněty kůže, abscesy, pneumonie, kolititidy, stomatitidy, perirektální záněty, sepse)
|
|
|
Eozinofily
|
název
|
eozinofilie
|
eozinopenie
|
|
referenční hodnota
|
> 0,450*109 eozinofilů/l periferní krve
|
< 1 %
|
|
|
příčiny vzniku
|
- alergická onemocnění
- dermatitidy
- tumory (lymfomy, melanomy, nádory mozku)
- paraziti (protozoa, červi)
- GIT nemoci (Crohnova choroba, ulcerózní kolitida, eozinofilní gastroenteritida)
- virové infekce
- jiné onemocnění (imunodeficience, chronické onemocnění ledvin, pneumonie)
- hypereozinofilní syndrom (onkologické onemocnění)
|
- infekce, kdy je zvýšeno uvolňování adrenalinu
- závažná zánětlivá reakce
- při virózách a během febrilních period
|
|
|
Bazofily
|
název
|
bazofilie
|
bazopenie
|
|
referenční hodnota
|
> 0,1*109 bazofilů/l periferní krve
|
|
|
|
příčiny vzniku
|
- alergické reakce (potravinová a léková alergie)
- infekce
- záněty (revmatická artritida, ulcerózní kolitida)
- jiné onemocnění: diabetes mellitus, mastocytóza, hemolytická anémie, karcinomy
- začínající menstruace
|
- ataka akutní revmatické horečky
- lobární pneumonie
- anafylaktoidní purpury
- chronická myeloidní leukémie
- tyreotoxikóza
- léčba kortikosteroidy, antihistaminiky
|
|
|
Lymfocyty
|
název
|
lymfocytóza
|
lymfopenie
|
|
referenční hodnota
|
> 4,0*109 všech lymfocytů/l
|
< 1,5*109 všech lymfocytů/l (pro dospělé)
|
|
|
příčiny vzniku
|
- virové infekce (viry hepatitidy A, B, C, HIV, zarděnky, EB viróza, herpes)
- bakteriální infekce (tuberkulóza, brucelóza)
- malignita (lymfomy, lymfatická leukémie)
- alergie
- endokrinologické příčiny (hypertyreóza, Basedowova choroba, Addisonova choroba)
|
- maligní onemocnění
- kolagenózy
- chemoterapie
- radioterapie
- kortikoterapie
- srdeční a ledvinné nedostatečnosti
- miliární tuberkulóza
|
|
|
Monocyty
|
název
|
monocytóza
|
monocytopenie
|
|
referenční hodnota
|
> 0,8*109 monocytů/l
|
< 0,1*109 monocytů/l
|
|
|
příčiny vzniku
|
- infekce (TBC, sepse, syfilis)
- hematologická onemocnění (některé druhy leukémií, myelodysplastický syndrom, některé neutropenie)
- jiná onemocnění (revmatoidní artritida, kolagenóza, zánětlivá onemocnění střeva, malignity)
|
- kortikoterapie
- aplastická anémie
- porucha zrání myelomonocytární linie krvetvorby
- vlasatobuněčná leukemie
|
|
4 Hematologické malignance
4.5 Testovací otázky
4.1 Myelodysplastický syndrom
4.2 Akutní leukémie
4.2.1 Akutní myeloidní leukemie
|
Příčiny AML
|
|
|
- radiační záření
- genetické změny (chromozomální přestavby)
- 5-10 let po léčbě alkylačními cytostatiky (cyklofosfamid, busulfan, cisplatina)
- 1-5 let po léčbě blokátory topoizomerázy II (etoposid, teniposid)
- chemikálie (benzen a jeho deriváty, herbicidy nebo pesticidy)
- hereditární onemocnění (Fanconiho anémie, Bloomův syndrom, Kostmanův syndrom, Downův nebo Klinefelterův syndrom)
|
|
|
Klinické příznaky
|
|
|
Z nedostatku erytrocytů
|
- anémie
- únava, slabost
|
|
Z nedostatku leukocytů
|
- nedostatečná imunita
- zvýšený výskyt infekcí horních cest dýchacích, pneumonie, stomatitidy, angíny se závažným průběhem, horečky bez prokázané infekce, opakované infekce i přes léčbu antibiotiky
- u 10% pacientů zvýšený počet leukocytů 100*109/l
|
|
Z nedostatku trombocytů
|
- trombocytopenie
- krvácení z nosu, dásní,
- petechie
- hyperplazie dásní
|
|
FAB subtyp
|
|
|
M0
|
akutní myeloblastická nediferencovaná – bez známek myeloidní diferenciace
|
|
M1
|
akutní myeloblastická bez vyzrávání – nediferencované myeloblasty
|
|
M2
|
akutní myeloblastická s vyzráváním – diferencované myeloblasty
|
|
M3
|
akutní promyelocytární – velké promyelocyty s hojnými granuly a Auerovými tyčkami
|
|
M4
|
akutní myelomonocytární – proliferace myeloidních a monocytárních prekurzorů
|
|
M5
|
akutní monocytární – přítomnost monoblastů (M5a) a monocytů (M5b)
|
|
M6
|
akutní erytroleukemie – přítomnost myeloblastů (M6a) a erytroblastů (M6b)
|
|
M7
|
akutní megakaryocytoblastická – přítomnost megakaryoblastů a megakaryocytů
|
4.2.2 Akutní lymfoblastová leukemie
4.3 Myeloproliferativní onemocnění
4.3.1 Chronická myeloidní leukemie
4.3.2 Pravá polycytemie (polycythaemia vera)
4.3.3 Esenciální (primární) trombocytémie
4.3.4 Chronická idiopatická myelofibróza
4.4 Lymfoproliferativní onemocnění
4.4.1 Maligní non-hodgkinské lymfomy
|
Typ vývojové linie
|
Jednotlivé typy NHL
|
Agresivita lymfomu
|
|
B-lymfocyt
|
Burkittův lymfom
Prekurzorový B-lymfoblastický lymfom
Plazmocelulární leukemie
Folikulární lymfom (velkobuněčný)
Difuzní velkobuněčný lymfom
Prolymfocytární B- leukemie
Mnohočetný myelom
Lymfom plášťové zóny
Vlasatobuněčná leukemie
Lymfom folikulárního centra (malobuněčný)
Extranodální MALT B-lymfom
Splenický lymfom marginální zóny
Lymfoplazmocytární lymfom
|
Velmi agresivní
Velmi agresivní
Velmi agresivní
Agresivní
Agresivní
Agresivní
Agresivní
Agresivní
Málo agresivní
Málo agresivní
Málo agresivní
Málo agresivní
Málo agresivní
|
|
T-lymfocyt
|
Sézaryho syndrom a Mycosis Fungoides
Anaplastický velkobuněčný lymfom
T a NK lymfatické leukemie
Chronická T-lymfatická leukemie
Periferní T-lymfomy
Intestinální T-lymfom
Angiocentrický lymfom
Prekurzorový T- lymfoblastický lymfom
T-lymfom/ HTLV1
|
Málo agresivní
Agresivní
Málo agresivní
Málo agresivní
Agresivní
Agresivní
Agresivní
Velmi agresivní
Velmi agresivní
|
|
Stádium
|
Projevy
|
|
Stádium I
|
zasažení jedné skupiny lymfatických uzlin (I)
nebo jednoho extralymfatického orgánu (I E)
|
|
Stádium II
|
zasažení dvou nebo více uzlin na jedné straně bránice (II)
nebo jedné či více skupin lymfatických uzlin a jednoho extralymfatického orgánu na jedné straně bránice (II E)
|
|
Stádium III
|
postižení lymfatických uzlin na obou stranách bránice (III)
nebo postižení lymfatických uzlin na obou stranách bránice a jednoho extralymfatického orgánu (III E), nebo sleziny (IIIS) nebo obojího (IIISE)
|
|
Stádium IV
|
postižení více extralymfatických orgánů (h-játra, i-plíce, b- kosti, m- kostní dřeň) s nebo bez postižení lymfatických uzlin
|
B lymfomy
4.4.2 Hodgkinův lymfom
|
Stádium
|
Systémové příznaky
|
|
Stádium I
|
zasažení jedné skupiny lymfatických uzlin (I)
nebo jednoho extralymfatického orgánu (I E)
|
|
Stádium II
|
zasažení dvou nebo více uzlin na jedné straně bránice (II)
nebo jedné či více skupin lymfatických uzlin a jednoho extralymfatického orgánu na jedné straně bránice (II E)
|
|
Stádium III
|
postižení lymfatických uzlin na obou stranách bránice (III)
nebo postižení lymfatických uzlin na obou stranách bránice a jednoho extralymfatického orgánu (III E), nebo sleziny (IIIS)
|
|
Stádium IV
|
postižení více extralymfatických orgánů bez ohledu na postižení uzlin
|